
本研究為描述中國華東地區腓骨肌萎縮症(Charcot-Marie-Tooth disease,CMT)患者的臨床、電生理及致病基因突變特點。探討臨床指標間的關係和基因型表型關 聯,並與國內外相似研究比較,總結異同。納入 2007-2013 年間上海華山醫院及福建醫科大學附屬第一醫院就診並接 受基因診斷的 CMT 患者,共計 148 個家係,入組家係的先證者由神經內科專病 醫生進行臨床及電生理評估。采用 FDS 評分及 CMTNS 評分評價患者疾病相關功能障礙。SPSS18.0 軟件用於統計分析。抽取枸櫞酸鈉抗凝外周靜脈血各 3 mL, 標準法提取基因組 DNA。MLPA 方法檢測 PMP22 基因擴增與缺失突變,Sanger 測序法檢測GJB1,MPZ,MFN2,GDAP-1 基因編碼區及其側翼部位的突變。分析基因型與臨床表型的關係。
結果表明
1.臨床特點
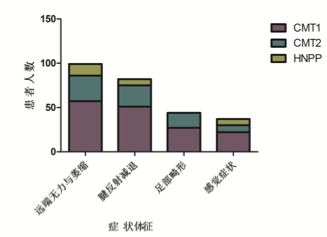
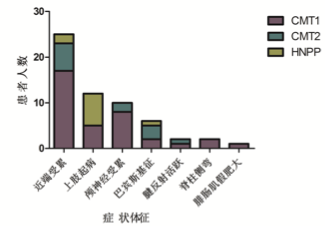
入組病例中,男女比例 1.4:1,平均就診年齡 24 歲。其中 CMT1 型患者比例 最高,其次是 CMT2 型及 HNPP,各型比例依次為 54.1%,28.8%,17.1%。入組 病例中,有陽性家族史占 45.0%,主要是常染色體顯性遺傳。58.5%患者 20 歲以 前起病,病情緩慢進展,致殘率低。CMT 常見的臨床表現包括四肢遠端肌無力 及萎縮,腱反射減退或消失,足部畸形,麻木或感覺減退。不常見的臨床表現有: 近端嚴重受累,上肢先起病,顱神經受累,病理征,腱反射亢進等。CMT1 型患 者中,CMTNS 評分與病程呈正相關,與 CMAP 呈負相關。與發病年齡及 MNCV 無顯著相關性。CMT2 型患者中未見上述指標間的顯著相關性。67%的 CMT 患 者肌酶輕度升高,符合神經源性損害特點。
入組病例常見和罕見臨床症狀體征
2 致病基因突變特點
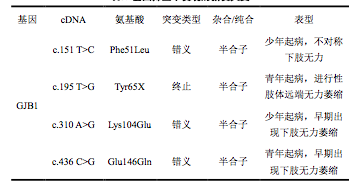
PMP22 基因擴增突變是最常見的突變類型(13.5%),其次是 PMP22 基因缺失 (11.5%)。GJB1 基因突變、MPZ 基因突變和 MFN2 基因突變各占 8.8%、2.0%和 0.7%,且 MPZ 基因突變多發生在 3 號外顯子。相關基因測序中共發現了 4 種新 突變類型,均為 GJB1 基因突變。在 GDAP-1 基因未找到突變。
基因篩查中發現的新發突變
3 基因型表型關係
CMT1A 型患者陽性家族史比例高(70%),常表現為經典的 CMT 症狀。正中神經 MNCV 普遍低於 30m/s,一半以上低於 20m/s。HNPP 患者症狀常反複發 作,亦有既往正常者,受壓後肢體麻木是最常見的主訴,肌電圖診斷與基因診斷 吻合度高。CMT1X 型患者症狀男性較女性嚴重,正中神經 MCV 輕度下降甚至 正常,多在 25-45m/s。CMT1B,分為早發-重症和晚發-輕症兩種表型。即使在同一位點突變,不同家係成員間的臨床表現可以有很大差異。
本研究患者隊列為國內目前最大的 CMT 患者隊列之一。入組患者中各臨 床亞型的分布與國外研究相似。患者的疾病相關功能障礙中國及其他亞洲人群的 PMP22 基因擴增突變頻率比歐美人群偏低。在對 CMT 患者行基因篩查前應先行肌電圖檢查。

